Snakemake: How to merge a variable number of files into one
Snakemake is a fantastic tool that has quickly become my go-to for bioinformatic workflows. I love its integration with conda, and I love not having to deal with job submissions because it plays so well with computer clusters.
Nevertheless, I find some of the “advanced” syntax a bit esoteric. Doing the straightforward 1-to-1 things is as easy as it gets: quick to write and run, super readable syntax, and there are plenty of examples online to learn this stuff, but I always end up spending far too much time taming the wildcards when I want to do anything more complex. There are some advanced workflows out there doing pretty impressive stuff, but in my opinion, there are not enough toy examples, which is ultimately how I always learn to do stuff. So, here’s one, in case you’re faced with a similar scenario:
I have a number of samples
samples, I have a variable number of files to download which are related to each sampledownloads_dict, I have the download URL for each sampleurls_dict. I want to download each file, and then merge them according to the sample they belong to.
What makes this tricky, is that the number of files to download and merge is variable across samples. Also, the name pattern for the downloaded files might not be directly inferrable from the sample name, so here’s where having some relationship dictionaries comes into play:
samples = ['sample_1', 'sample_2', 'sample_3']
downloads_dict = {'sample_1': ['download_1a', 'download_1b'],
'sample_2': ['download_2a'],
'sample_3': ['download_3a', 'download_3b', 'download_3c']}
urls_dict = {'download_1a': 'hi, its download 1a',
'download_1b': 'hi, its download 1b',
'download_2a': 'hi, its download 2a',
'download_3a': 'hi, its download 3a',
'download_3b': 'hi, its download 3b',
'download_3c': 'hi, its download 3c'}
rule all:
input:
expand('{sample}.merged.txt', sample=samples)
rule mock_download:
output:
'{download}.faa'
params:
url = lambda wc: urls_dict[wc.download]
shell:
'echo {params.url} > {output}'
rule merge:
input:
lambda wc: expand('{download}.faa', download=downloads_dict[wc.sample])
output:
'{sample}.merged.txt'
shell:
'cat {input} > {output}'
The DAG for this looks like:
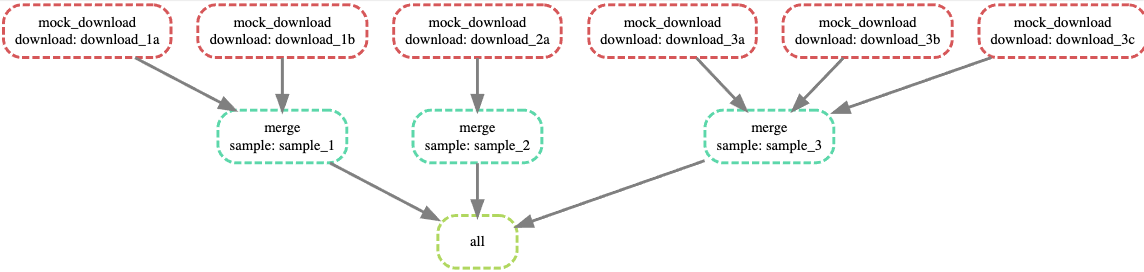
You’ll notice my “download” is actually just echo, but it very well could be curl, I just wanted it to be self-contained for the sake of this example. This is obviously a silly toy example, but I ultimately used something very similar to this to download fasta files through MGnify’s API and then merge them to further process them. 👌
Thanks to @maxgmarin, who showed me how to do this originally!